We have found 100,000 cells per assay to be sufficient for enriching target loci for multiple histone modifications, transcription factors, and transcription cofactors. However, one can increase the number of cells to 250,000 per assay without scaling up beads, antibodies, or buffers.
We have shown that our CUT&RUN Assay Kit #86652 works with as few as 5,000-10,000 cells for histone modifications and 10,000-20,000 cells for transcription factors and cofactors (see images).
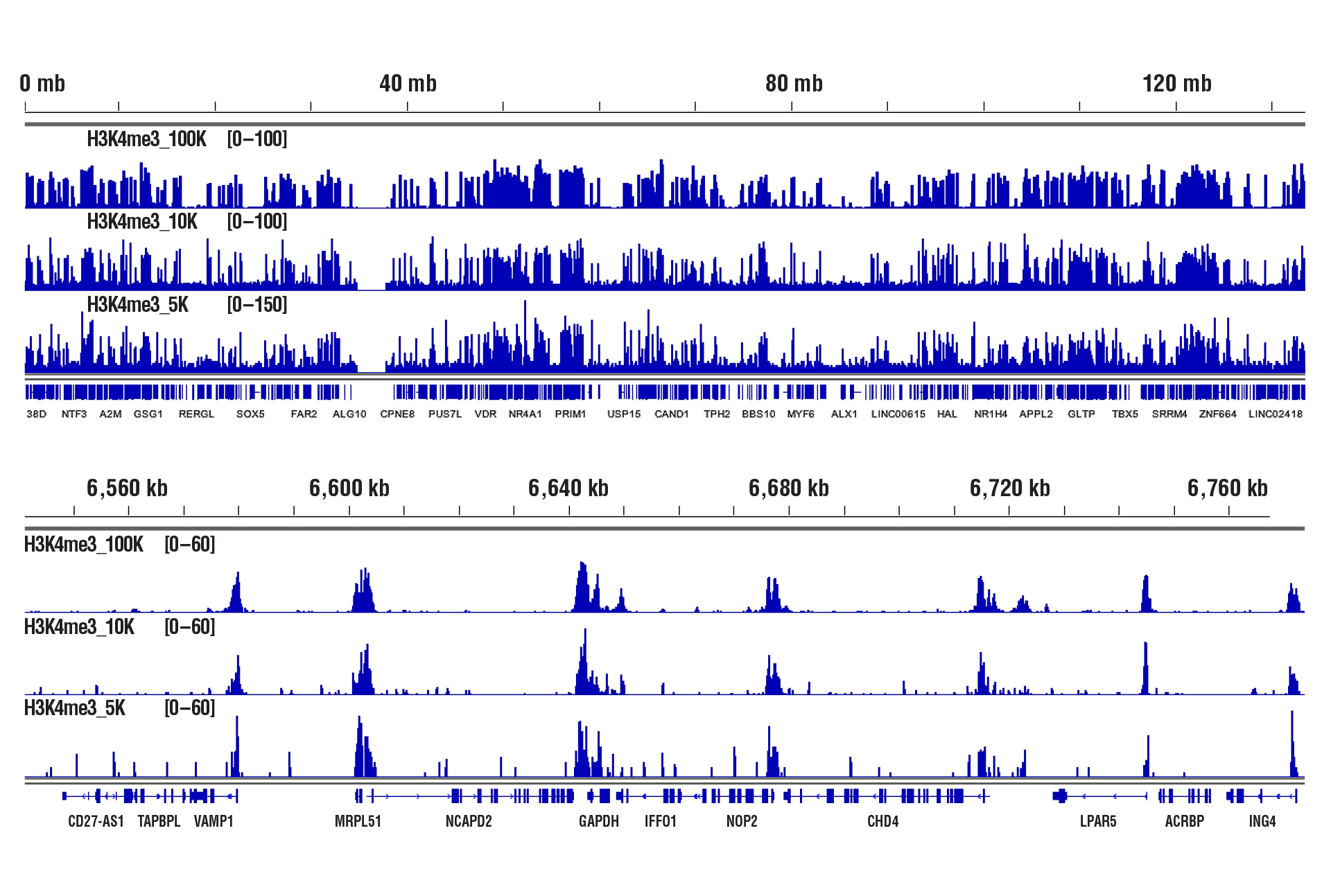
CUT&RUN was performed with 100,000, 10,000, or 5,000 HCT 116 cells (as indicated) and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel shows binding across chromosome 12, while the lower panel shows enrichment around the GAPDH gene, a known target of H3K4me3.
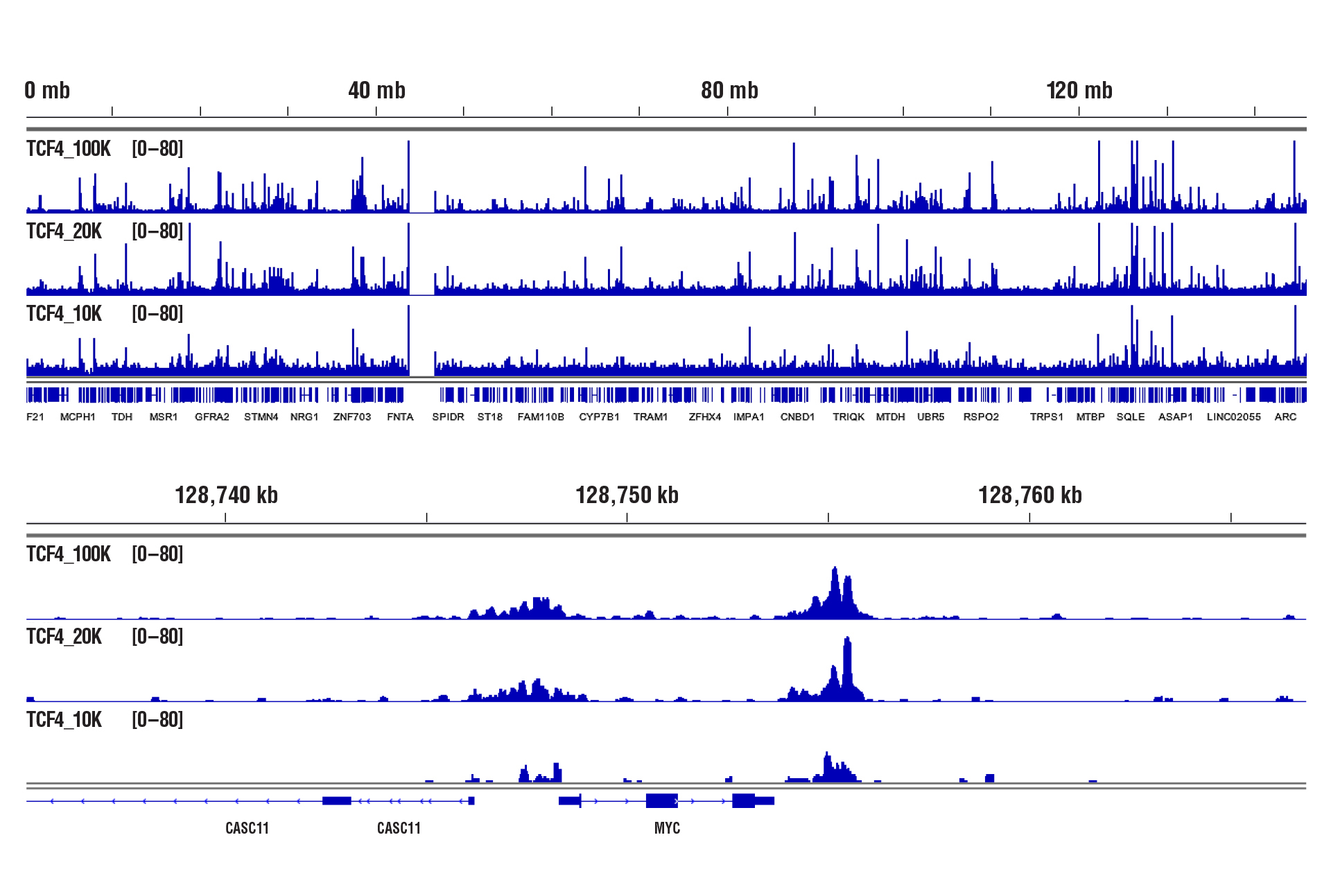
CUT&RUN was performed with 100,000, 20,000, or 10,000 HCT 116 cells (as indicated) and TCF4/TCF7L2 (C48H11) Rabbit mAb #2569, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel shows binding across chromosome 8, while the lower panel shows enrichment around the MYC gene, a known target of TCF4.
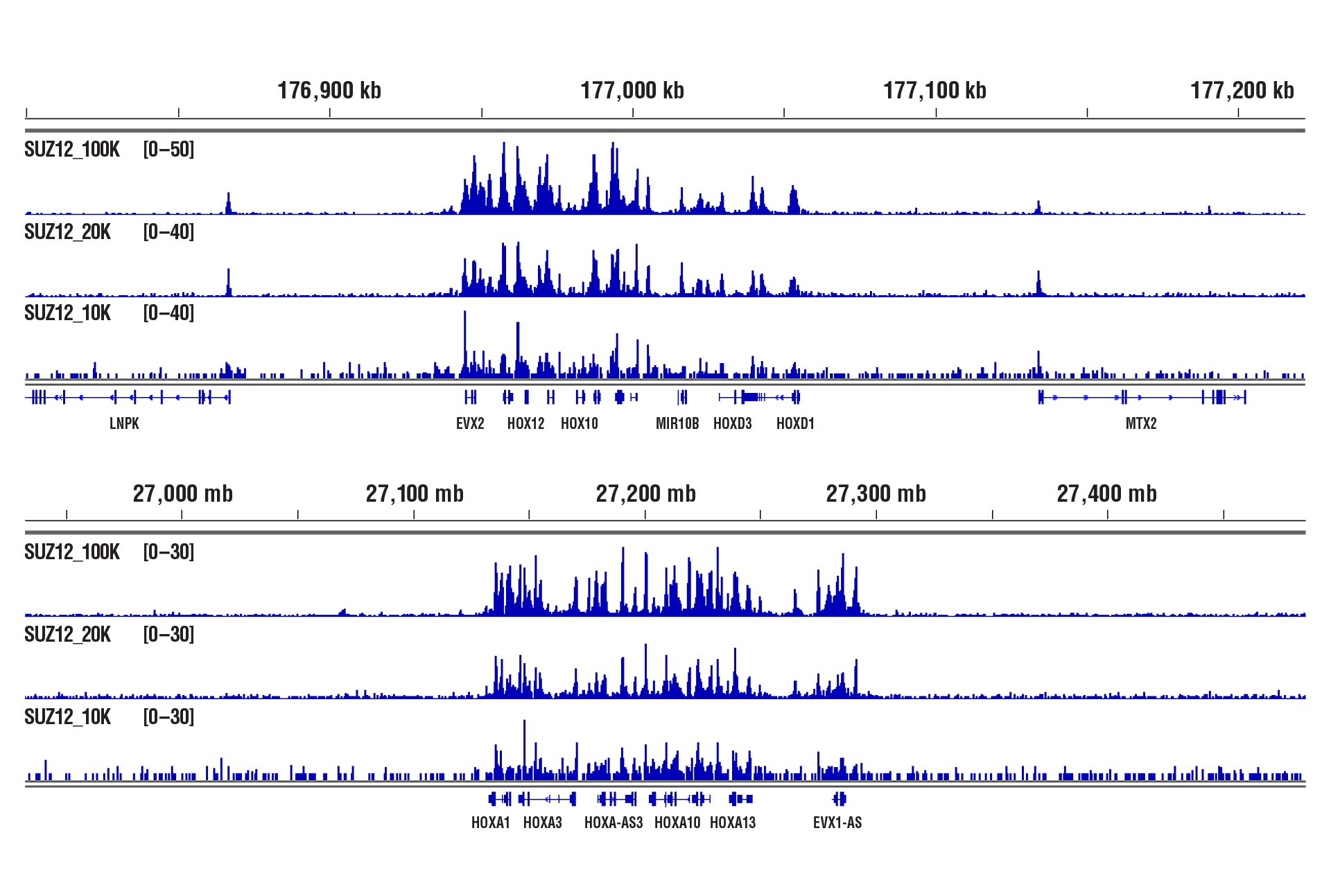
CUT&RUN was performed with 100,000, 20,000, or 10,000 NCCIT cells (as indicated) and SUZ12 (D39F6) XP® Rabbit mAb #3737, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figure shows binding across HoxD (upper panel) and HoxA (lower panel) gene clusters, known targets of SUZ12.
We have performed CUT&RUN on dozens of cultured adherent and suspension cell lines, both human and mouse. So far we have not noticed any significant difference in performance between these cell lines.
With adherent cell lines, one first needs to detach the cells from the dish. We recommend using trypsin to detach the cells. Alternatively, you can use accutase, but the resulting cell pellet doesn't pack as well during centrifugation, so you tend to lose cells during the wash steps. Suspension cells are much easier. Just count the cells using a hemacytometer or auto cell counter and then collect the number of cells you need. We don't observe a significant difference in assay performance with adherent versus suspension cells.
Yes. Cell fixation is recommended when the cells are very fragile (easily lysed during experiments), when the cellular signaling pathway is sensitive to the Concanavalin A beads used for cell binding, when the target protein binds weakly to DNA, or when you hope to freeze the cell pellets for future use. Using a light fixation protocol (0.1% formaldehyde for 2 min) is critical to the success of performing CUT&RUN with fixed cells. A reverse crosslinking step is also required at the end of the assay to remove protein from DNA. We find that live cells generally have better performance than fixed cells in the CUT&RUN assay, therefore, we recommend using live cells if possible, unless fixation is necessary in above mentioned scenarios (see image).
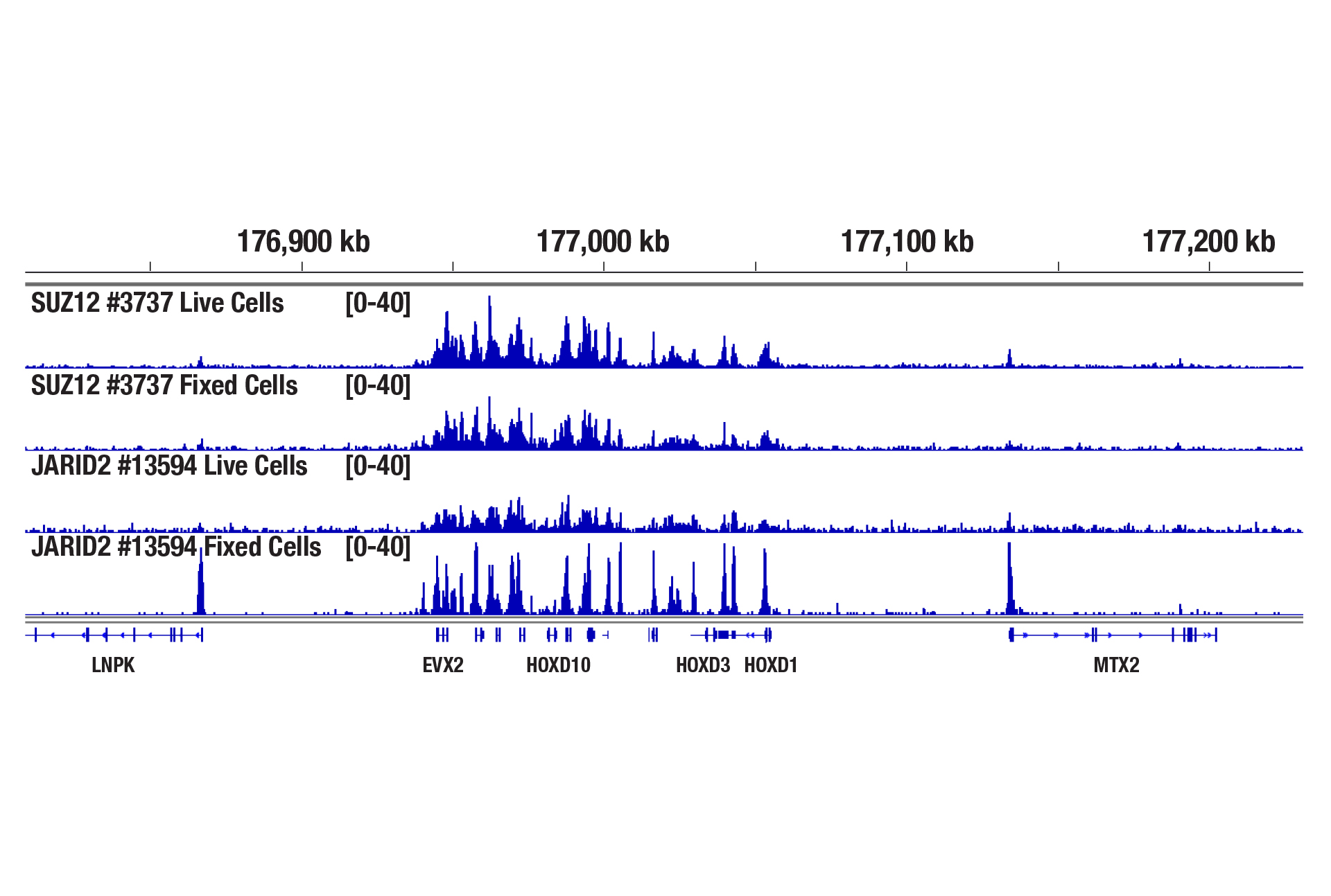
CUT&RUN was performed with 100,000 live or fixed NCCIT cells and SUZ12 (D39F6) XP® Rabbit mAb #3737 or JARID2 (D6M9X) Rabbit mAb #13594, using the CUT&RUN Assay Kit. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figure shows binding of SUZ12 and JARID2 across the HoxD gene cluster. Fixation is not required for SUZ12 (a core component of PRC2 complex) but increases the enrichment for JARID2 (an accessory component of PRC2 complex).
Yes. We have shown that our CUT&RUN Assay Kit #86652 works with 100,000 human CD8+ live T cells with antibodies against epigenetic regulators and transcription factors (see image).
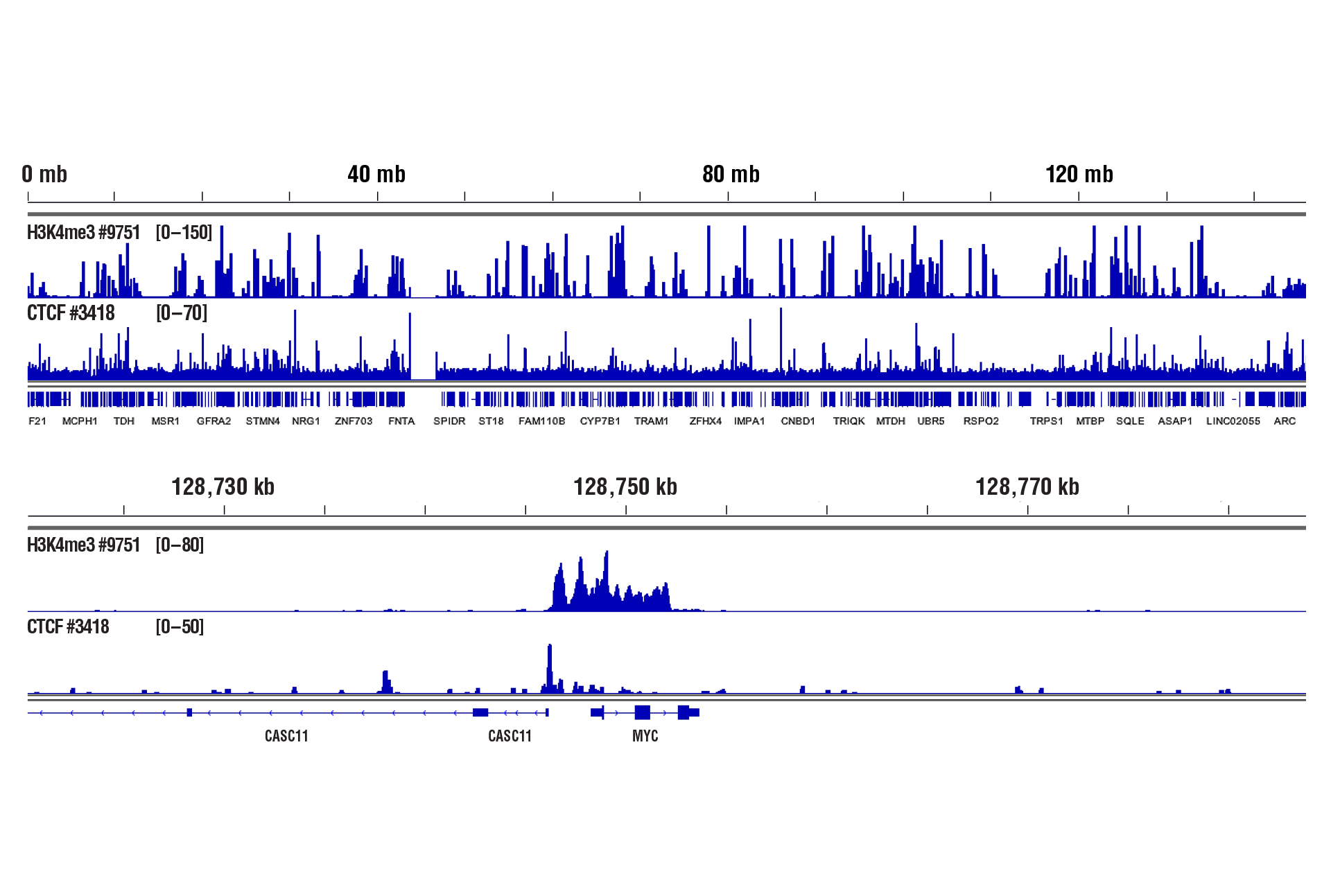
CUT&RUN was performed with 100,000 live human CD8+ T cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 or CTCF (D31H2) XP® Rabbit mAb #3418, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel shows binding across chromosome 8, while the lower panel shows enrichment around the MYC gene, a known target of H3K4me3 and CTCF.
Yes. We have shown that our CUT&RUN Assay Kit #86652 works with mouse liver, mouse brain, and mouse heart tissues with as little as 1 mg of tissue for each CUT&RUN reaction. Fresh tissue samples work with histone modifications and high abundant DNA binding factors like CTCF. However, for optimal results with transcription factors, cofactors, and difficult tissue types, a light to medium fixation should be performed (0.1% formaldehyde, 2-10 min) and slightly more starting material (2.5-5 mg) should be used (see images). Fixed tissues can be frozen for up to 6 months before use in the CUT&RUN assay.
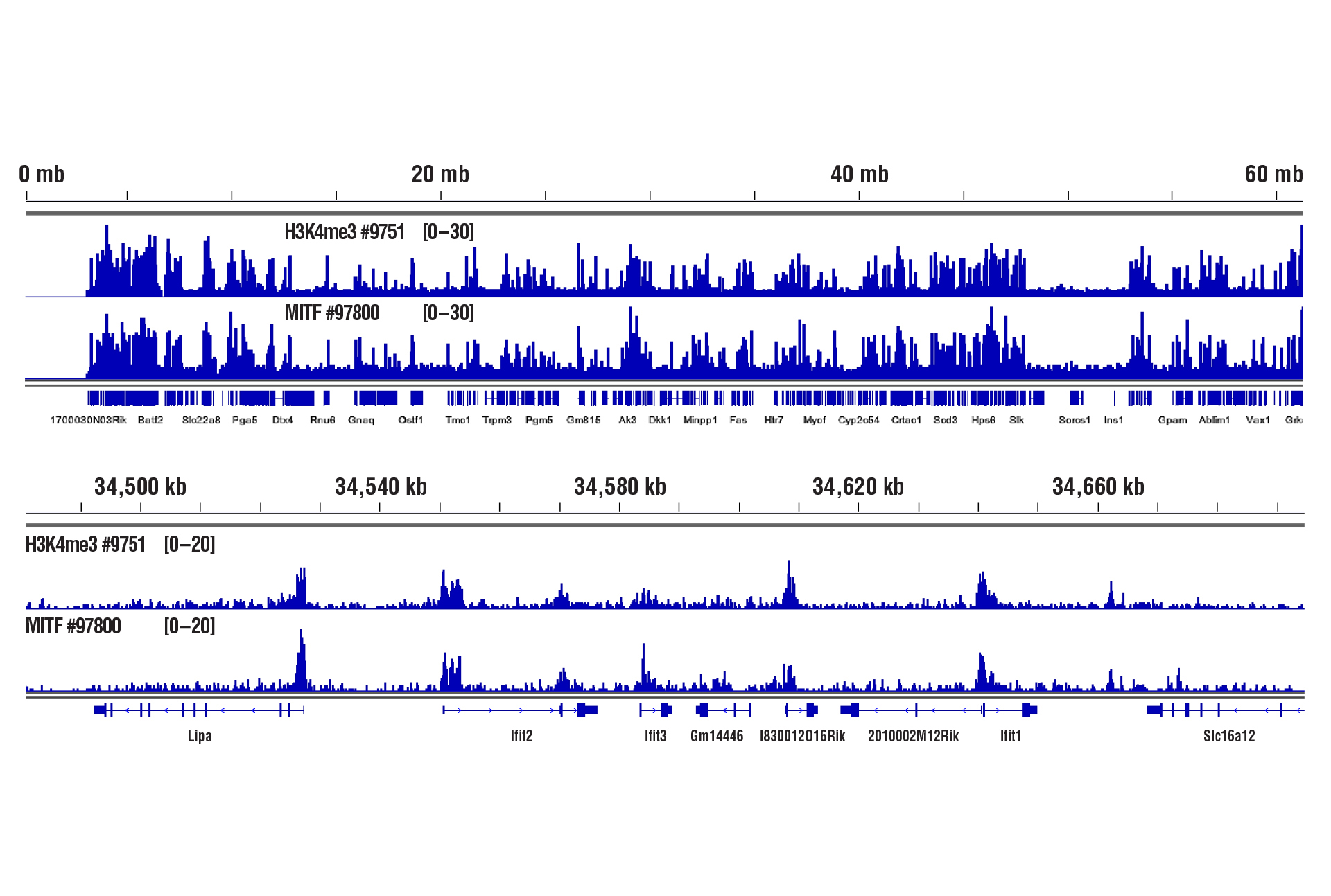
CUT&RUN was performed with 1 mg of lightly fixed mouse heart tissue (0.1% formaldehyde, 2 min) and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 or MITF (D3B4T) Rabbit mAb #97800, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel shows binding of H3K4me3 and MITF across chromosome 19, while the lower panel shows binding around the Ifit2 gene.
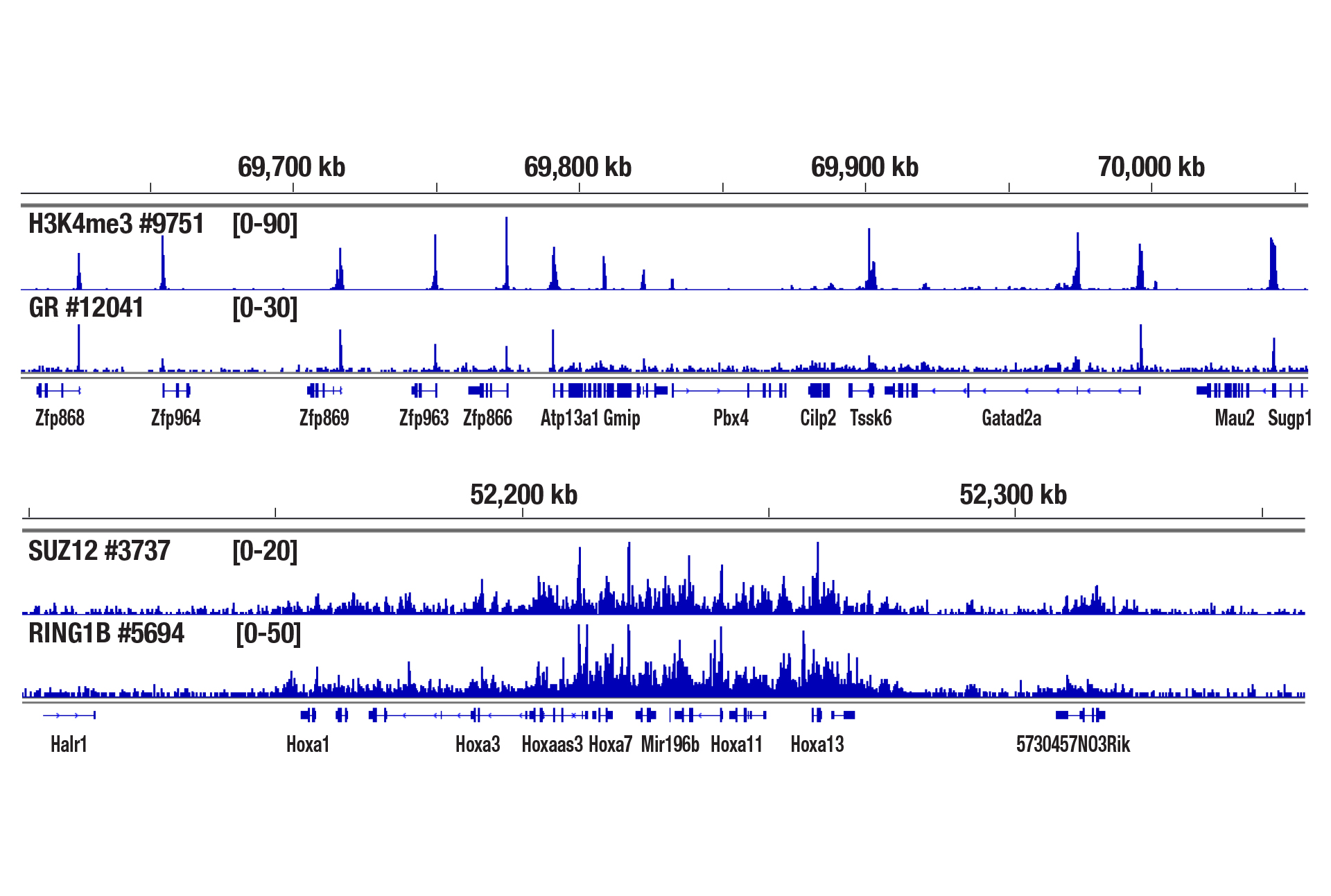
CUT&RUN was performed with 2.5 mg of medium-fixed mouse liver tissue (0.1% formaldehyde, 10 min) and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, Glucocorticoid Receptor (D6H2L) XP® Rabbit mAb #12041, SUZ12 (D39F6) XP® Rabbit mAb #3737, or RING1B (D22F2) XP® Rabbit mAb #5694, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel shows binding of H3K4me3 and Glucocorticoid Receptor around the Zfp866 gene, while the lower panel shows binding of SUZ12 and RING1B across the HoxA gene cluster.
We have not tested our CUT&RUN kit using plant cells. However, Xiao-Yu Zheng et al. (2019; PMID30719569) describes a method for pre-treating plant tissues and preparing the nuclei for CUT&RUN. Our kit can be used downstream of this nuclei preparation protocol. In this case, digitonin may not be required for cell permeabilization during the CUT&RUN assay, but it may help further permeabilize the cell membrane if the plant cells are not 100% homogenous.
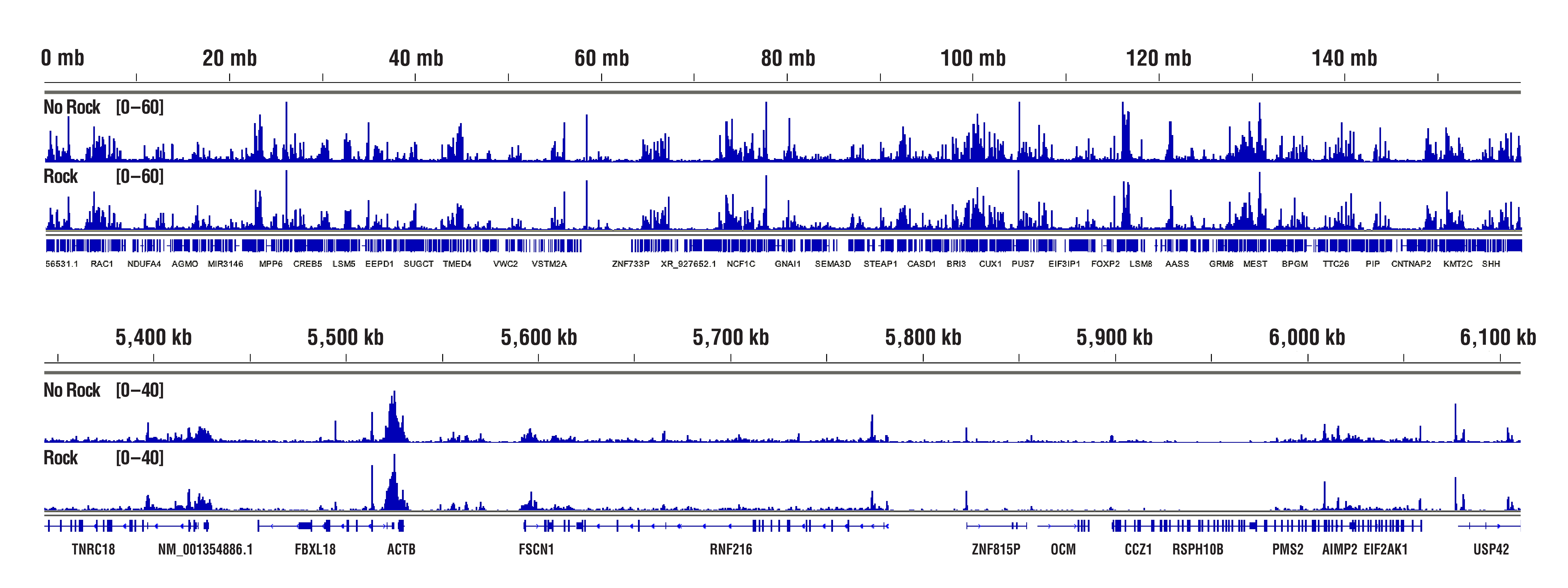
The Concanavalin A beads can clump together rather easily, especially when the cells become lysed. Make sure your cells are healthy and be sure to treat them very gently during the cell wash. We also recommend limiting the room temperature incubation time of the cell suspension with the Concanavalin A beads to 5 minutes. Another option instead of rotating or rocking the tubes during the incubations for cell-bead binding, antibody binding, and pAG-MNase enzyme binding is to just let the tubes rest at the appropriate incubation temperatures. This seems to help to mitigate the beads from clumping and does not have any adverse effects on the final results (see figures below). With all that being said, our internal testing has shown that bead clumping does not negatively affect the final results of CUT&RUN experiments.
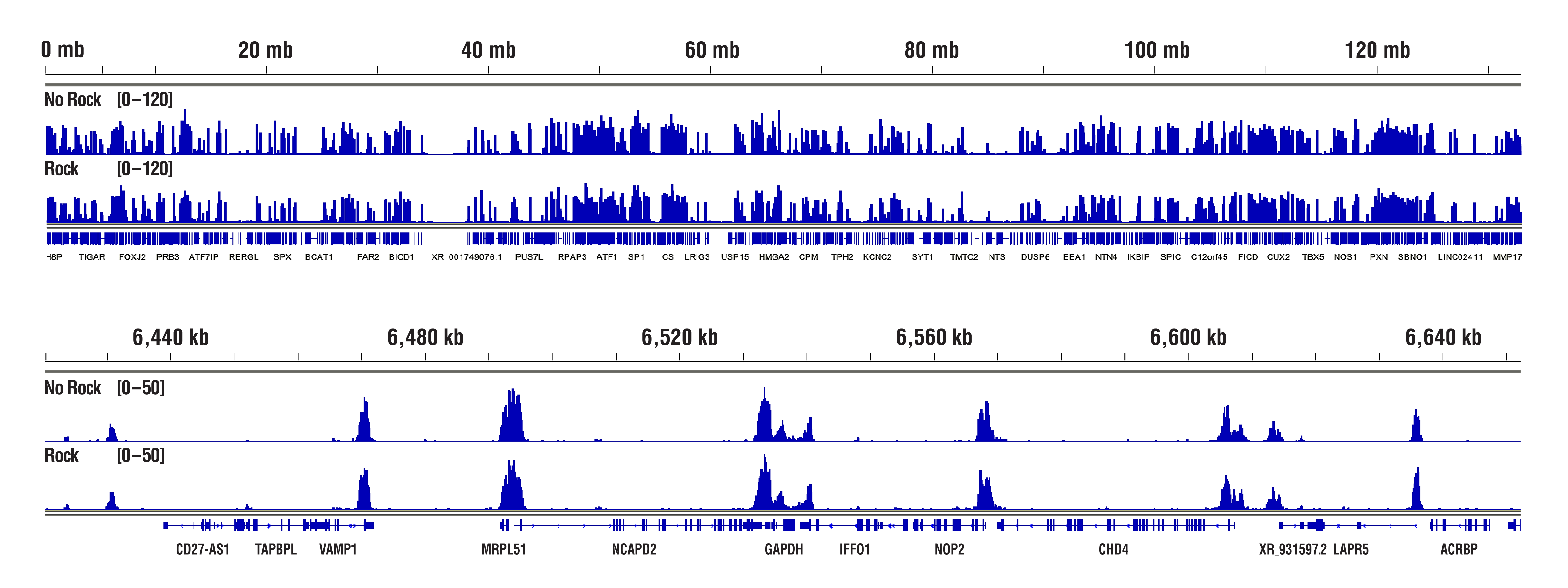
H3K4me3 data highlighting comparable results in "Not Rocked" vs "Rocked" samples. CUT&RUN was performed with 20,000 HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, using CUT&RUN Assay Kit #86652. Samples were either left sitting still (No Rock) or rocked on nutator (Rock) at appropriate temperatures during incubations for cell-bead binding, antibody binding, and pAG-MNase enzyme binding. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 12 (upper), including GAPDH (lower), a known target gene of H3K4me3. No significant difference in signal strength or binding pattern was observed between the No Rock and Rock protocols.
Rpb1 data highlighting comparable results in "Not Rocked" vs "Rocked" samples. CUT&RUN was performed with 100,000 HCT 116 cells and Phospho-Rpb1 CTD (Ser2) (E1Z3G) Rabbit mAb #13499, using CUT&RUN Assay Kit #86652. Samples were either left sitting still (No Rock) or rocked on a nutator (Rock) at appropriate temperatures during incubations for cell-bead binding, antibody binding, and pAG-MNase enzyme binding. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 7 (upper), including ACTB (lower), a known target gene of Phospho-Rpb1. No significant difference in signal strength or binding pattern was observed between the No Rock and Rock protocols.
Our optimized digestion conditions for the CUT&RUN assay work well across multiple cell lines, both adherent and suspension. In addition, we haven't observed significant changes in chromatin digestion when increasing or decreasing either the amount of pAG-MNase added or the time of digestion. Our observations are similar to those published by Peter Skene, et al. (2018; PMID29651053). That said, the digestion temperature does matter. We find that digestion at 4°C can significantly increase signal strength with an acceptable background for transcription factors and cofactors compared to digestion at 0°C (see images).
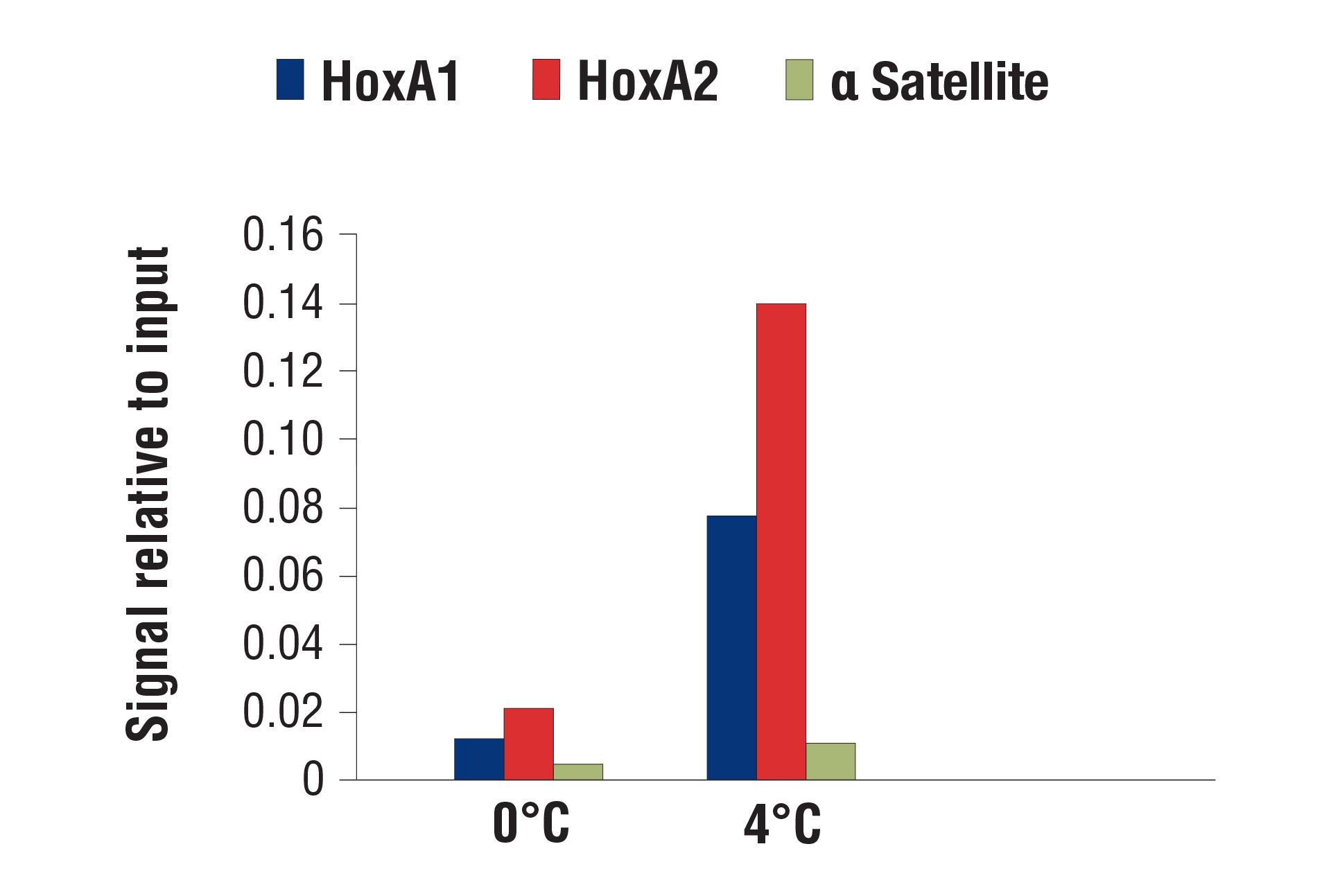
CUT&RUN was performed with NCCIT cells and SUZ12 (D39F6) XP® Rabbit mAb #3737, using CUT&RUN Assay Kit #86652. DNA was digested at either 0°C or 4°C as indicated. The enriched DNA was quantified by real-time PCR using SimpleChIP® Human HoxA1 Intron 1 Primers #7707, SimpleChIP® Human HoxA2 Promoter Primers #5517, and SimpleChIP® Human α Satellite Repeat Primers #4486. The amount of immunoprecipitated DNA in each sample is represented as signal relative to the total amount of input chromatin, which is equivalent to one.
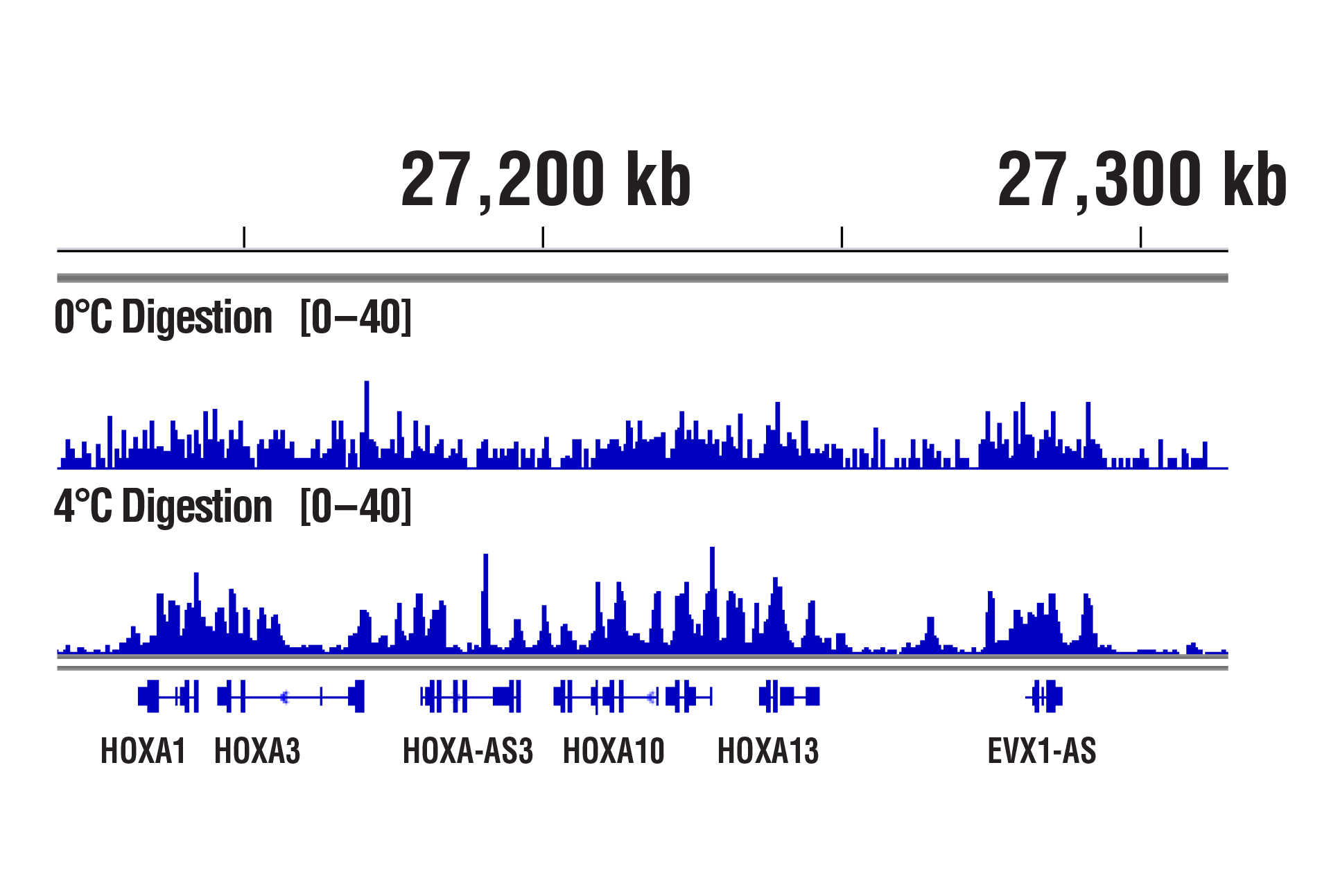
CUT&RUN was performed with NCCIT cells and SUZ12 (D39F6) XP® Rabbit mAb #3737, using CUT&RUN Assay Kit #86652. DNA was digested at either 0°C or 4°C as indicated. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figure shows binding across HoxA genes.
Yes, the digitonin needs to be present in all of the buffers during the entire experiment because the membrane permeabilization caused by digitonin is reversible. Removing or lowering the amount of digitonin in the buffers may interfere with both the antibody and pAG-MNase entering the nuclei. Cells are more sensitive to digitonin concentration rather than the digitonin treatment time. If you find that the recommended amount of digitonin is leading to extensive lysis of your cells, you can optimize the concentration of digitonin in your assay as described in Appendix A in our CUT&RUN kit protocol.
Our CUT&RUN kit is optimized for use with whole cells and works with a large number of cell lines, both adherent and suspension. While it is not typically necessary to pre-isolate cell nuclei for CUT&RUN, our kit should work with pre-isolated nuclei because the Concanavalin A beads bind to glycoproteins on the surface of both cells and nuclei. Pre-isolation of cell nuclei should only be necessary for cells that are not efficiently permeabilized by digitonin, such as yeast or plant cells that both contain cell walls.
No. We don't observe any bias toward euchromatin or heterochromatin in our assays. In CUT&RUN, the target-specific antibody recruits the pAG-MNase and directs digestion to chromatin that is directly adjacent to the antibody, whether that be in euchromatin or heterochromatin. In this case, the active tethering of pAG-MNase to the chromatin allows for digestion to occur even in less accessible heterochromatin. Our CUT&RUN Assay Kit #86652 has been shown to work well with antibodies against active, accessible euchromatic histone modifications (see Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751) and activating transcription factors (see NF-kB p65 [D14E12] XP® Rabbit mAb #8242)), as well as repressive, inaccessible heterochromatic histone modifications (see Tri-Methyl-Histone H3 [Lys27] [C36B11] #9733) and polycomb repressor complex proteins that are associated with inactive heterochromatin (see EZH2 [D2C9] XP® Rabbit mAb #5246 and RING1B [D22F2] XP® Rabbit mAb #5694) (see images).
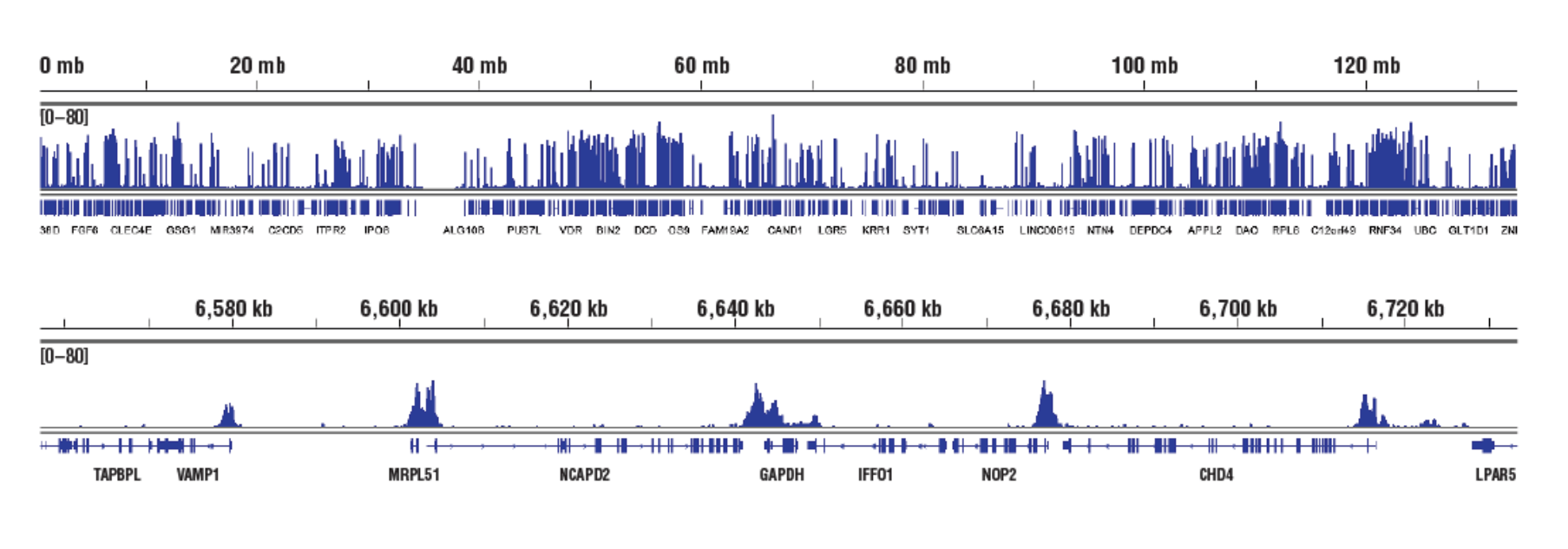
Accessible Chromatin, Target: H3K4me3: CUT&RUN was performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 12 (upper), including GAPDH (lower), a known target gene of H3K4me3.
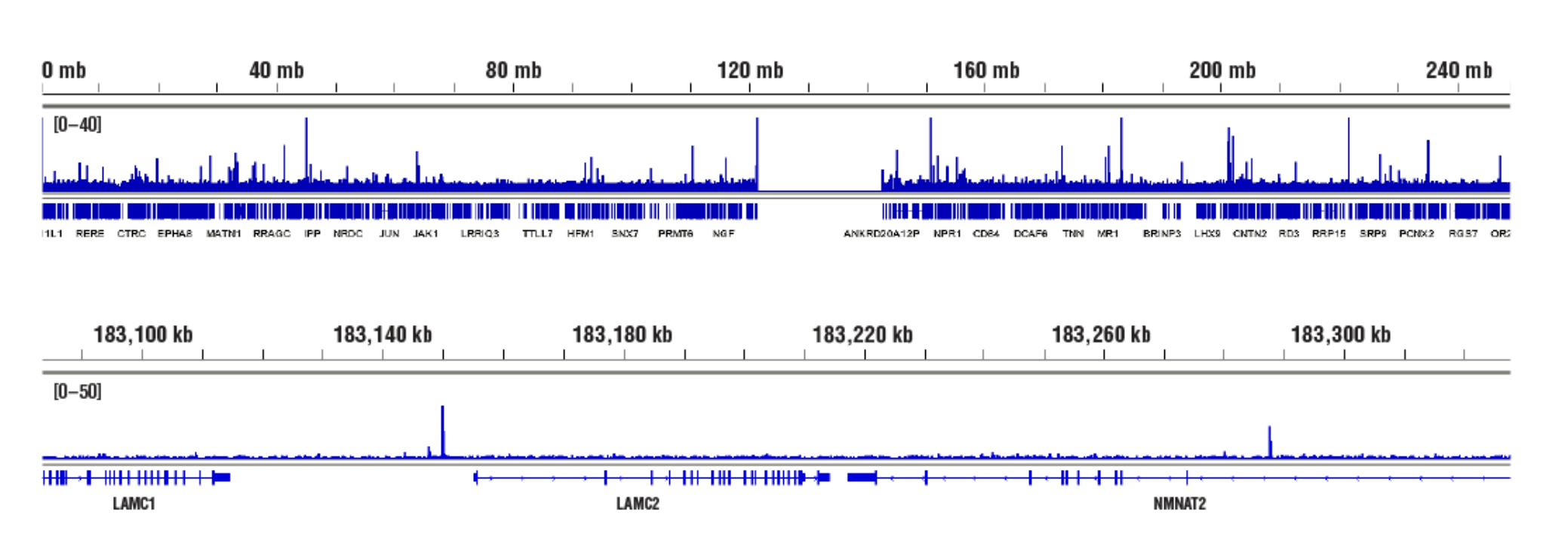
Accessible Chromatin, Target: NF-κB p65: CUT&RUN was performed with HeLa cells treated with hTNF-α #8902 (30 ng/ml, 1 hr) and NF-κB p65 (D14E12) XP® Rabbit mAb #8242, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 1 (upper), including LAMC2 (lower), a known target gene of NF-κB p65
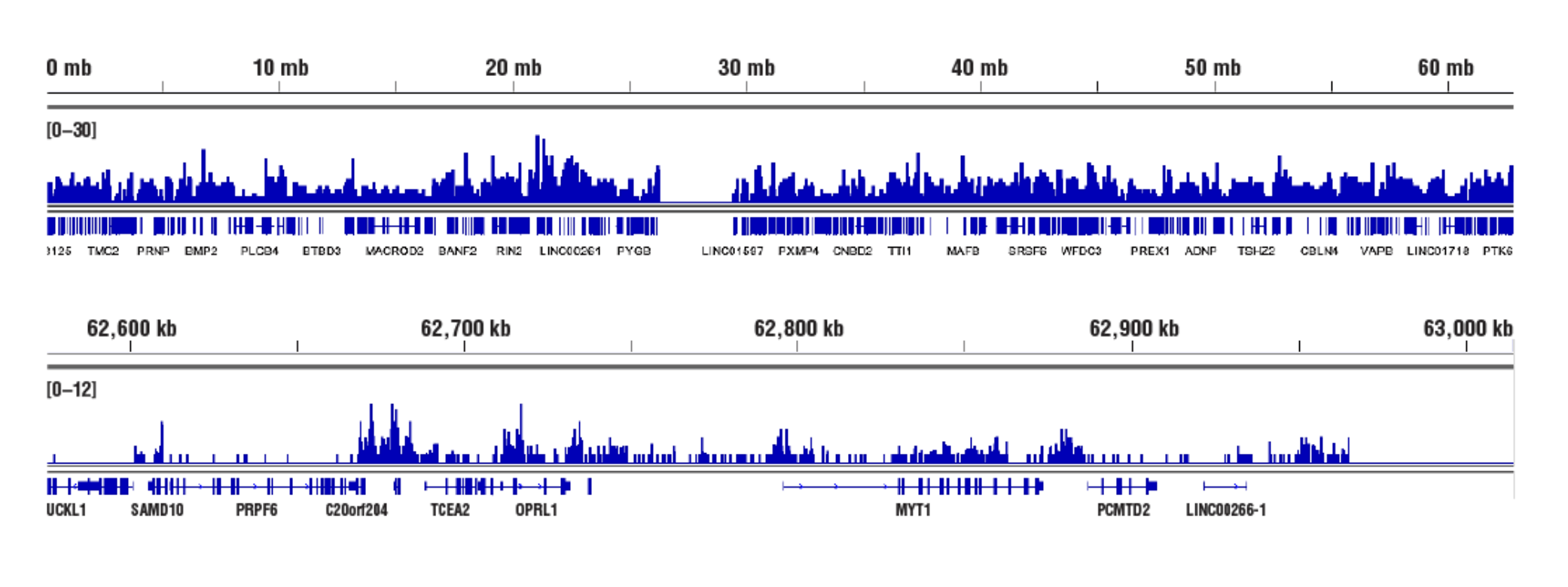
Inaccessible Heterochromatin, Target: Tri-Methyl-Histone H3: CUT&RUN was performed with HeLa cells and Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb #9733, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 20 (upper), including TCEA2 gene (lower).
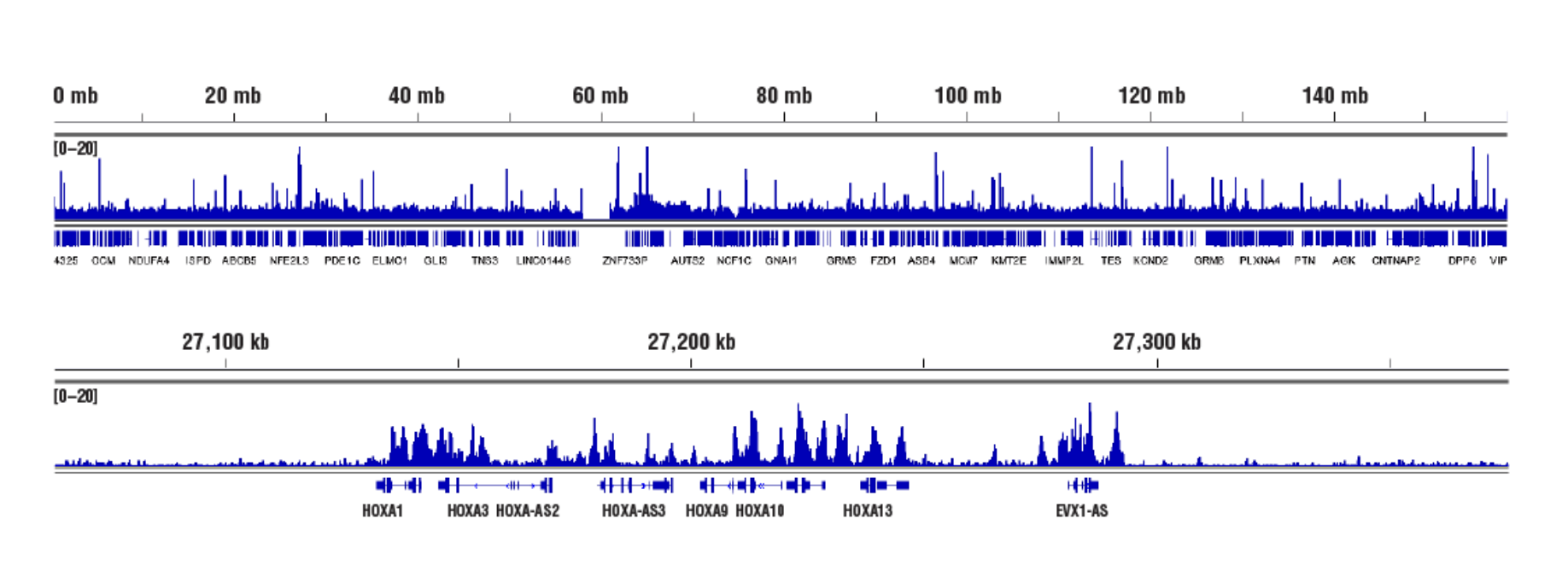
Inaccessible Heterochromatin, Target: Ezh2: CUT&RUN was performed with NCCIT cells and Ezh2 (D2C9) XP® Rabbit mAb #5246, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The figures show binding across chromosome 7 (upper), including HoxA genes (lower), a known cluster of EZH2 target genes.
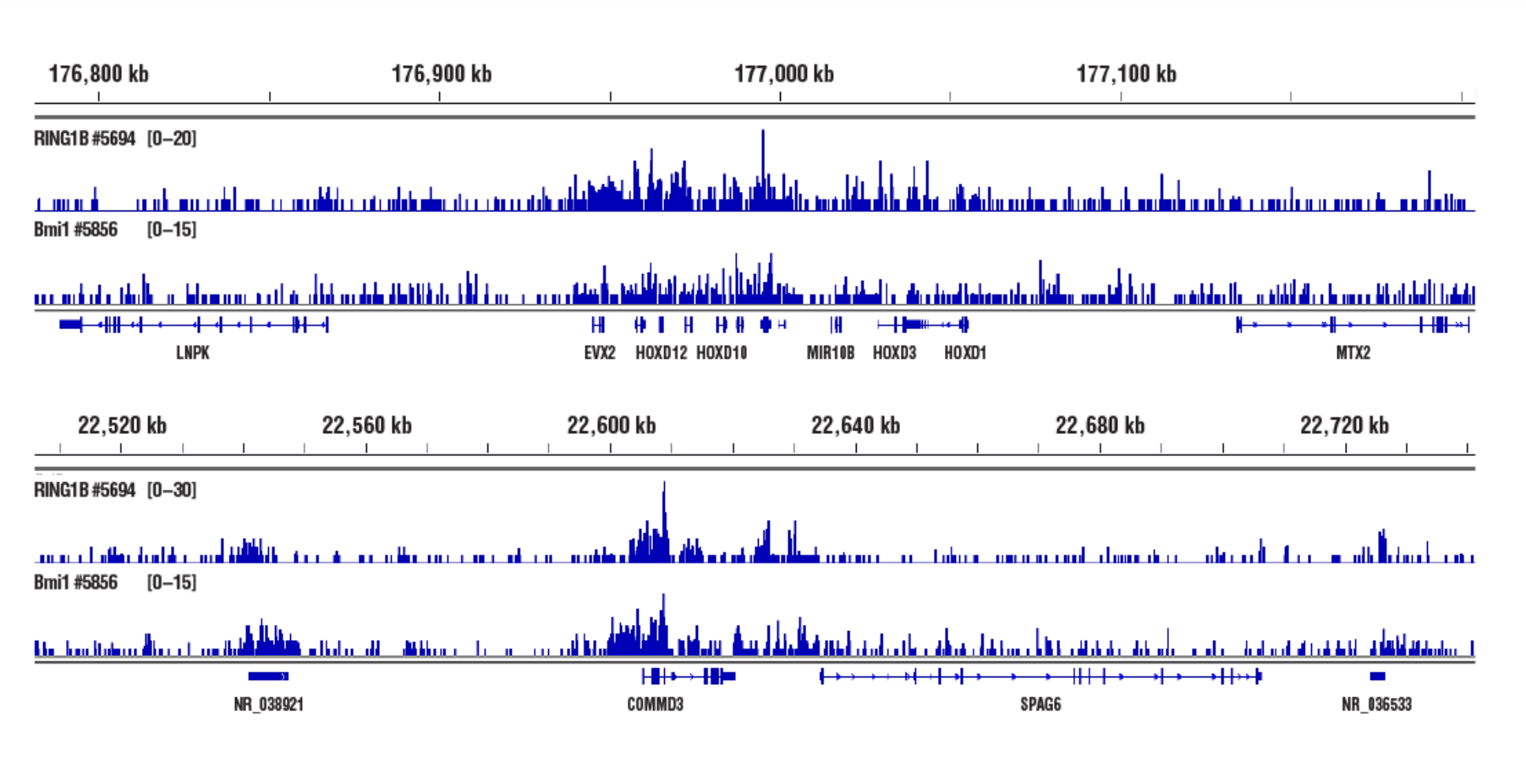
Inaccessible Heterochromatin, Target: RING1B: CUT&RUN was performed with NCCIT cells and either RING1B (D22F2) XP® Rabbit mAb #5694 or Bmi1 (D42B3) Rabbit mAb #5856, using CUT&RUN Assay Kit #86652. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. RING1B and Bmi1 are both PRC1 components. The figures show binding across HOXD (upper) and COMMD3 (lower) genes.
Whether performing CUT&RUN with downstream qPCR or NG-seq analysis, we always recommend using a CUT&RUN-validated positive control antibody like our Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 and a negative control antibody like our Rabbit (DA1E) mAb IgG XP® Isotype Control Antibody #66362. These antibodies can be paired with our SimpleChIP® Human RPL30 Exon 3 Primers #7014 or SimpleChIP® Mouse RPL30 Intron 2 Primers #7015 to show that your CUT&RUN assay is working regardless of the performance of your desired target-specific antibody. All of these controls are included in our CUT&RUN Assay Kit #86652 (see images).
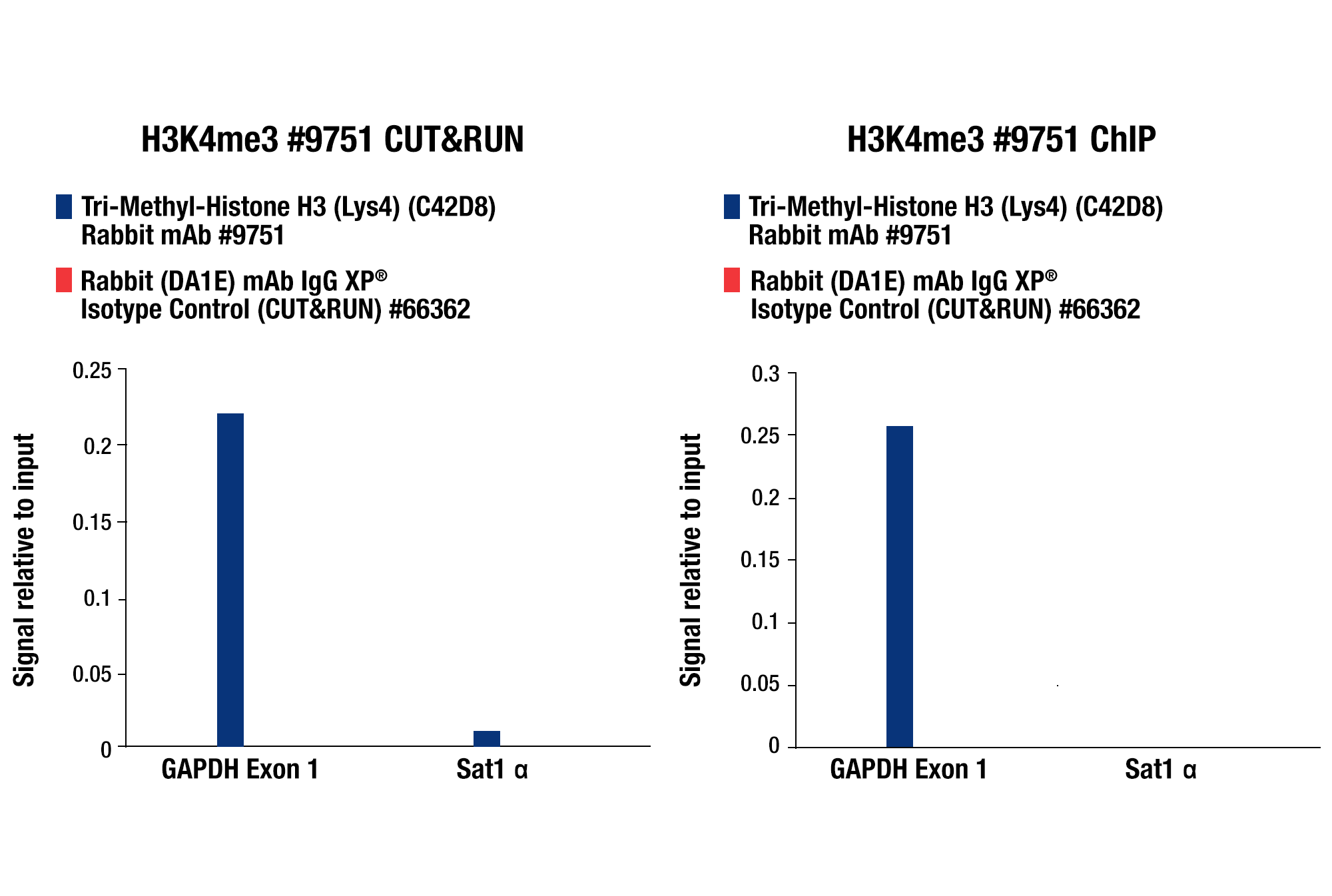
CUT&RUN qPCR Control Data: CUT&RUN and ChIP assays were performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751 or Rabbit (DA1E) mAb IgG XP® Isotype Control (CUT&RUN) #66362, using this CUT&RUN Assay Kit #86652 (left panel) or the SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005 (right panel). The enriched DNA was quantified by real-time PCR using SimpleChIP® Human GAPDH Exon 1 Primers #5516 and SimpleChIP® Human α Satellite Repeat Primers #4486. The amount of immunoprecipitated DNA in each sample is represented as signal relative to the total amount of input chromatin, which is equivalent to one.
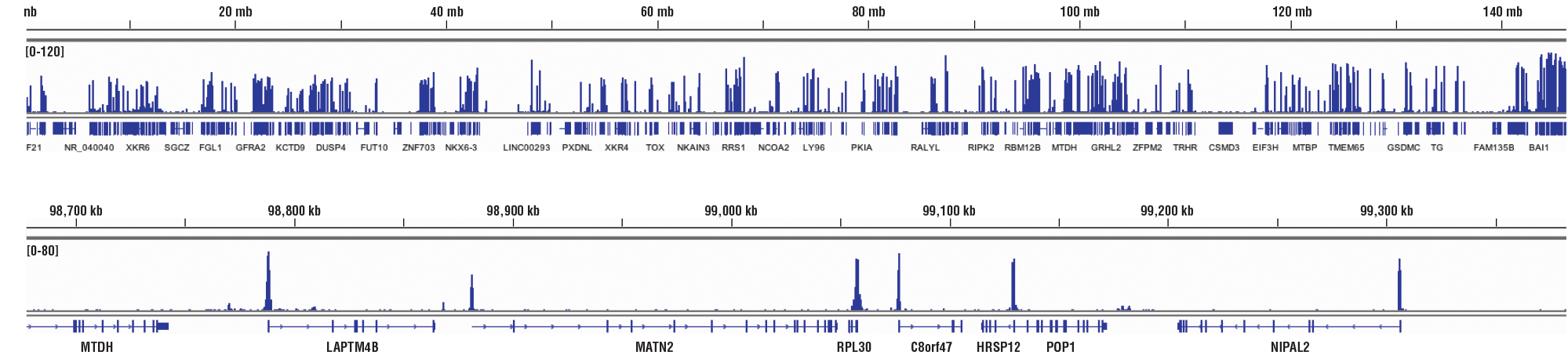
CUT&RUN NGS Control Data: CUT&RUN and ChIP assays were performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751, using this CUT&RUN Assay Kit #86652 or the SimpleChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) #9005. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The upper panel compares enrichment of H3K4me3 across chromosome 12, while the lower panel compares enrichment at the GAPDH gene, a known target of H3K4me3. The input tracks are from the CUT&RUN input sample.
We recommend starting with a ChIP- or ChIP-seq-validated antibody; however, we have found that not all ChIP-validated antibodies work in the CUT&RUN assay. Alternatively, Peter J. Skene et al. (2017; PMID28079019) suggest that you may want to try an antibody validated for an immunofluorescence assay, as the antibody binding in CUT&RUN takes place in an intact nuclear environment, resembling conditions for antibody binding in immunofluorescence assays.
Some protocols utilize contaminating E. coli DNA in the pAG-MNase for sample normalization. However, this can become problematic when switching lots of pAG-MNase, because each lot of enzyme will have differing amounts of contaminating DNA. Instead, we recommend using a separate yeast spike-in DNA sample. We provide a yeast spike-in DNA sample with our CUT&RUN Assay Kit #86652 and our CUT&RUN pAG-MNase and Spike-In DNA #40366 for which every lot is tested and optimized for CUT&RUN. This yeast spike-in DNA provides better lot-to-lot consistency for your normalization efforts.
A specified amount of pre-fragmented yeast genomic DNA is added to each CUT&RUN reaction after the digestion and prior to DNA purification. This spike-in component allows you to normalize for differences in DNA purification, qPCR, library preparation and sequencing efficiencies between samples. The protocol included in our CUT&RUN kit provides detailed instructions for the spike-in DNA control for both qPCR and NG-seq. It is important to note that significantly different amounts of spike-in DNA are required for qPCR vs. NG-seq, so please read the protocol carefully.
When using antibodies against histone modifications, we typically see fragment sizes as small as 150 bp (size of a mono-nucleosome) and quite often see a nucleosomal ladder of DNA fragment sizes (i.e. 150, 300, 450, 600 bp). When using antibodies against transcription factors, we see a range of DNA fragment sizes, with the majority being greater than 35 bp (see images).
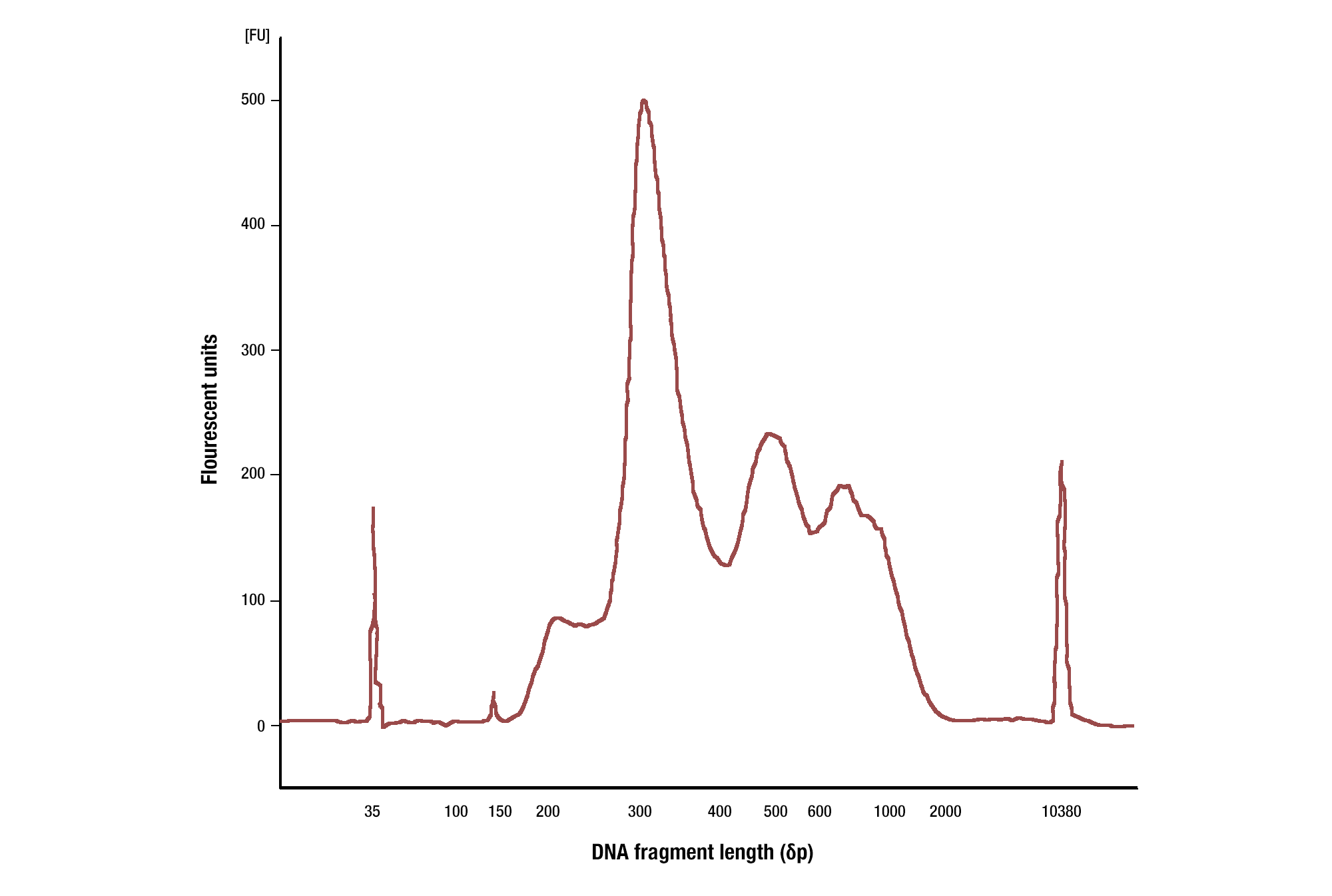
Target: H3K4me3: CUT&RUN assays were performed with HCT 116 cells and Tri-Methyl-Histone H3 (Lys4) (C42D8) Rabbit mAb #9751. DNA Libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. The size of the DNA fragments in the library were analyzed using an Agilent Bioanalyzer. The adaptor and barcode sequences added to the library during construction account for 140 bp in fragment length. As shown, excised DNA is highly enriched for mononucleosomes (peak around 300 bp).
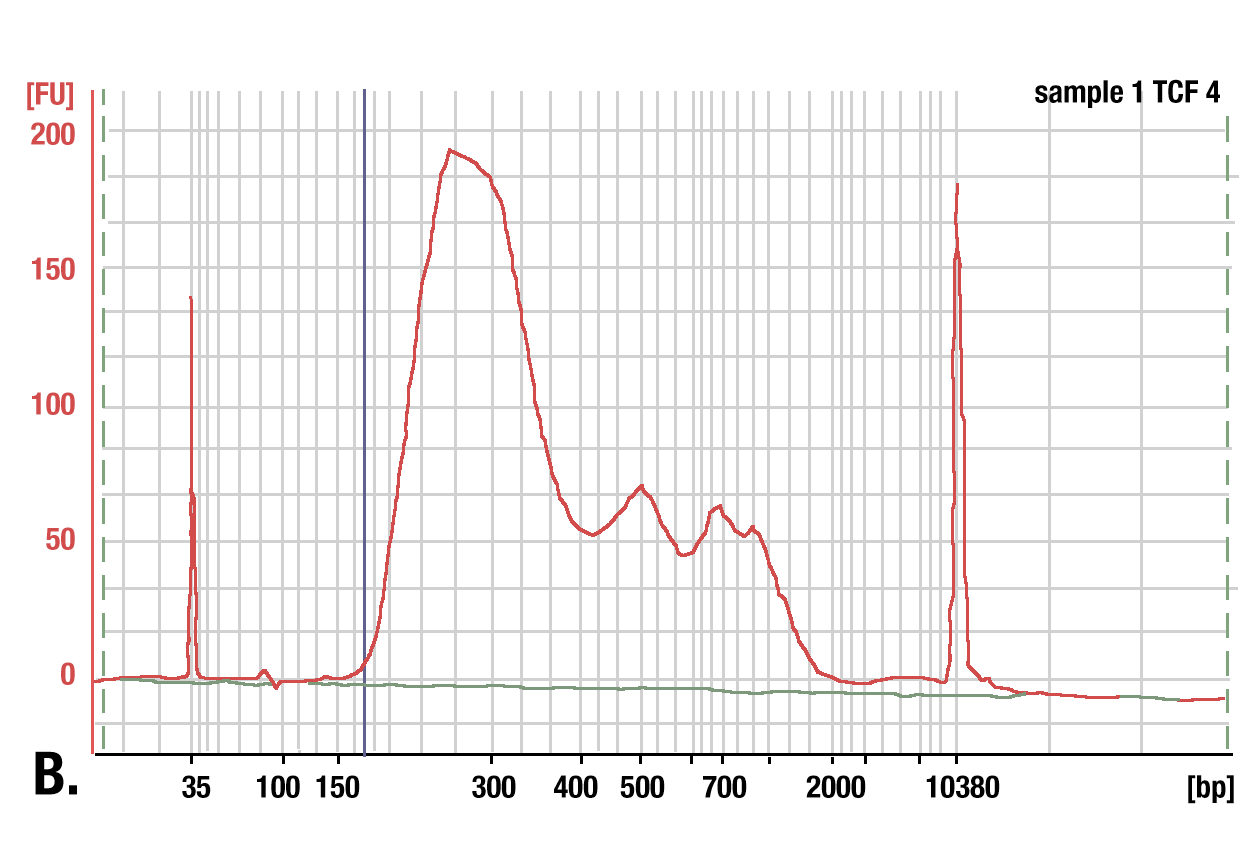
Target: TCF4/TCF7L2: DNA was purified using phenol/chloroform extraction followed by ethanol precipitation from a CUT&RUN assay performed using TCF4/TCF7L2 (C48H11) Rabbit mAb #2569. The size of the DNA fragments in the library was analyzed using a Bioanalyzer (Agilent Technologies). The adaptor and barcode sequences added to the library during construction account for 140 bp in fragment length.
We recommend using our DNA Purification Buffers and Spin Columns (ChIP & CUT&RUN) #14209 for purifying your CUT&RUN DNA. These spin columns provide an easy and inexpensive method for purifying CUT&RUN DNA. In a side-by-side comparison, our DNA spin columns effectively recovered DNA fragments greater than, or equal to, 35 bp. While phenol-chloroform extraction followed by ethanol precipitation did more effectively recover smaller DNA fragments, when analyzing DNA libraries made from both samples, we found that the DNA columns captured approximately 98% of the total CUT&RUN DNA fragments. Approximately 2% of the DNA fragments found in the phenol-chloroform-extracted and ethanol-precipitated sample were less than 35 bp in length. Therefore, our DNA spin columns provide a simple, inexpensive, and robust method for purifying your CUT&RUN DNA fragments. Please note that not all DNA spin columns capture the same range of DNA fragment sizes, so if you use DNA spin columns from a different vendor, you will want to determine the minimum size of DNA fragment retained by those columns, as a higher fragment size cutoff can negatively impact your results (see image).
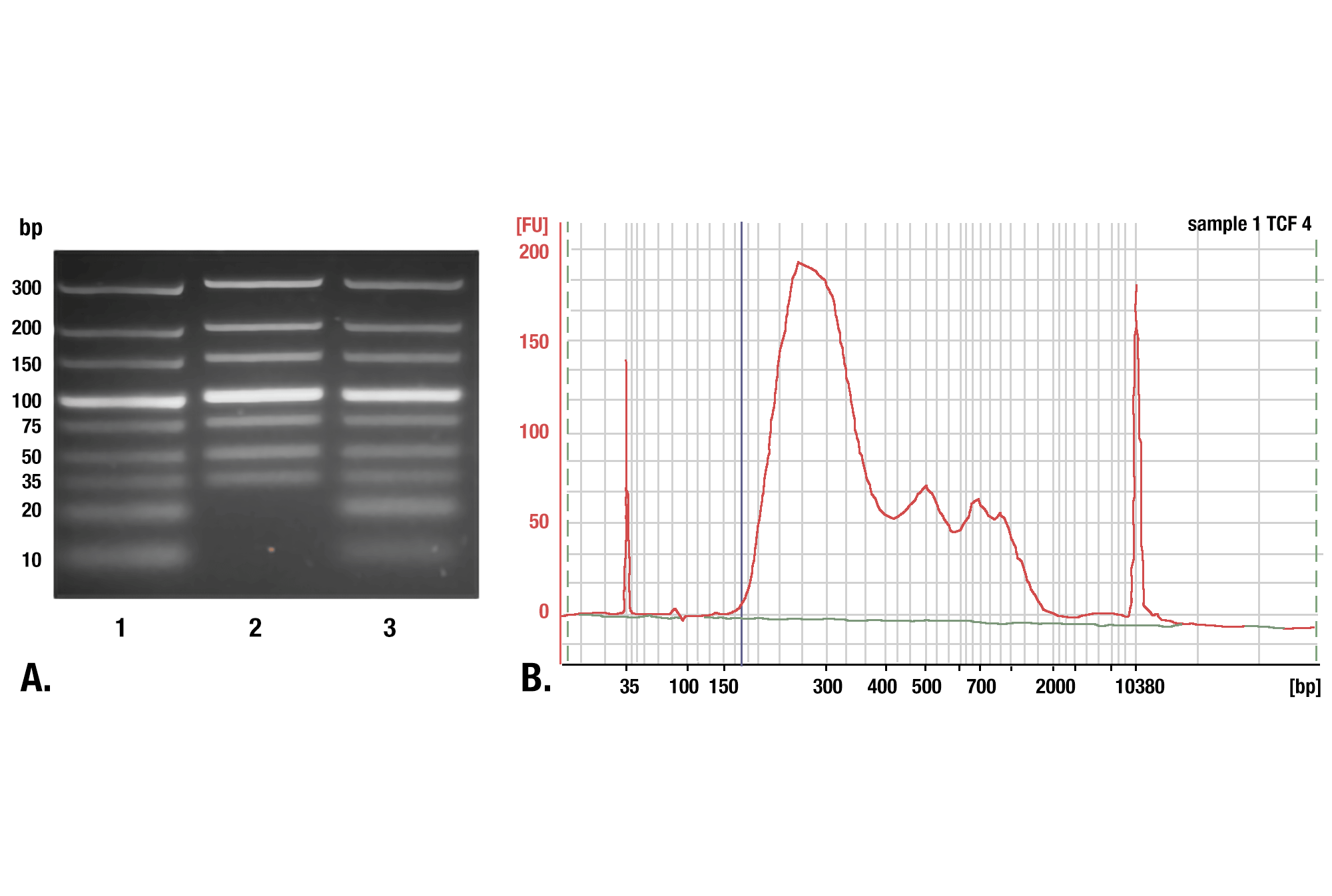
Phenol/Chloroform Extraction vs. Spin Columns for DNA Purification: Comparison of DNA purification using spin columns or phenol/chloroform extraction followed by ethanol precipitation. (A) A low range DNA ladder mix (lane 1, unpurified) was purified using either DNA Purification Buffers and Spin Columns (ChIP, CUT&RUN) #14209 (lane 2) or phenol/chloroform extraction followed by ethanol precipitation (lane 3) and separated by electrophoresis on a 4% agarose gel. As shown, phenol/chloroform followed by ethanol precipitation efficiently recovers all DNA fragment sizes, while DNA spin columns recover DNA fragments ≥35 bp. (B) DNA was purified using phenol/chloroform extraction followed by ethanol precipitation from a CUT&RUN assay performed using TCF4/TCF7L2 (C48H11) Rabbit mAb #2569. The size of the DNA fragments in the library was analyzed using a Bioanalyzer (Agilent Technologies). The adaptor and barcode sequences added to the library during construction account for 140 bp in fragment length. Therefore, starting 35 bp DNA fragments would be 175 bp in length after library preparation (indicated with blue vertical line in figure). As shown, less than 2% of the total CUT&RUN enriched DNA fragments are less than 175 bp (starting length of 35 bp), suggesting that DNA purification spin columns are sufficient for the capture of >98% of the total CUT&RUN DNA fragments.
Our CUT&RUN kit is compatible with downstream qPCR analysis. The protocol with the kit provides instructions and reagents for generating an input DNA sample that can be used to quantify your CUT&RUN DNA by qPCR. For qPCR analysis of CUT&RUN DNA, we recommend designing primers that generate amplicons of 60 to 80 bp in length.
You can QC your CUT&RUN assay prior to NG-seq by performing qPCR analysis of your enriched DNA. Our CUT&RUN kit is compatible with downstream qPCR analysis.
When performing CUT&RUN with downstream qPCR analysis, one can use a normal IgG antibody enriched sample as the negative control and calculate the target-specific antibody enrichment as fold over IgG background. However, the IgG sample typically contains extremely small amounts of DNA and is not a good indicator for how well your target-specific antibody is working in the assay. Therefore, we recommend generating and using an input chromatin DNA sample. How to generate an input chromatin DNA sample is described in Section V of our CUT&RUN kit protocol. This way, you can express the target-specific antibody DNA enrichment as percent of the total input DNA and directly determine how well the target-specific antibody is working in the assay.
The Bioanalyzer requires 5 to 10 ng of DNA for optimal analysis of DNA fragment sizes. However, a successful CUT&RUN experiment can often yield less than 1 ng of DNA from 100,000 starting cells. This low yield is most common for transcription factor and cofactor analyses. We have found that this low yield is okay and still provides robust sequencing data. In these instances, we recommend that you proceed with your DNA squencing library preparation and then analyze your DNA library using a Bioanalyzer (typical concentration of DNA library is 10 to 40 ng/μl). Instead of relying on DNA yield as a QC measurement of your CUT&RUN experiment, we recommend analyzing your CUT&RUN experiment by qPCR to look at enrichment of a few known target genes prior to performing NG-seq.
This depends on the sensitivity of the DNA library prep kit. The typical yield for CUT&RUN DNA is 0.6 to 6 ng per reaction. Our DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795 will work with starting DNA as low as 0.5 ng. You can also increase the number of PCR cycles up to 20 to increase amplification of the library if the starting amount is too low.
We recommend using our DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795 for making DNA libraries with CUT&RUN or ChIP DNA. This kit should be paired with our Multiplex Oligos for Illumina (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538 or Multiplex Oligos for Illumina (Single Index Primers) (ChIP-seq, CUT&RUN) #29580.
No, we don't recommend performing size selection during the library preparation for CUT&RUN or ChIP-seq. Based on our experience and feedback from customers, size selection can significantly decrease both the yield and diversity of the DNA library. If you are concerned about large DNA fragments >1 kb, then simply decrease the extension time when amplifying the adaptor-ligated DNA sample during the libarary preparation. We find that reducing the extension time to 10 to 15 seconds greatly reduces amplification of large fragments > 1kb, while being sufficient for amplification of smaller and more desirable DNA fragments during the CUT&RUN library preparation.
When performing CUT&RUN with downstream NG-seq analysis, one can use a normal IgG antibody enriched sample as the negative control. However, we have seen and heard from other scientists that normal IgG antibodies generate very low diversity DNA libraries and can generate what looks to be specific enrichment of genomic regions in the CUT&RUN assay, complicating your NG-seq data analysis. Instead, we recommend generating and using an input chromatin DNA sample as described in Section V of our CUT&RUN kit protocol. This input DNA sample generates a more diverse DNA library and works better as a negative control for analysis by NG-seq.
If a sonicator is not available and downstream NG-seq analysis is desired, please use the Digestion of CUT&RUN Input DNA by MNase protocol found on our Protocols page. If a sonicator is not available and qPCR analysis is desired, the un-fragmented input DNA can be used for qPCR analysis; however, un-fragmented input DNA should be purified using phenol/chloroform extraction and ethanol precipitation because the size of un-fragmented input DNA is too large to be purified using DNA spin columns. Alternatively, enzymatically digested input DNA can be used for qPCR analysis as well.
This decision was based on our analysis of hundreds of CUT&RUN samples. We found that a sequencing depth lower than 3 million generated too few peaks and that the number of peaks increased with deeper sequencing. If desired, one can increase the sequencing depth to 10 million reads per sample. The duplication rate of sequencing reads significantly increases if the sequencing depth is >15 million reads per sample (see image).
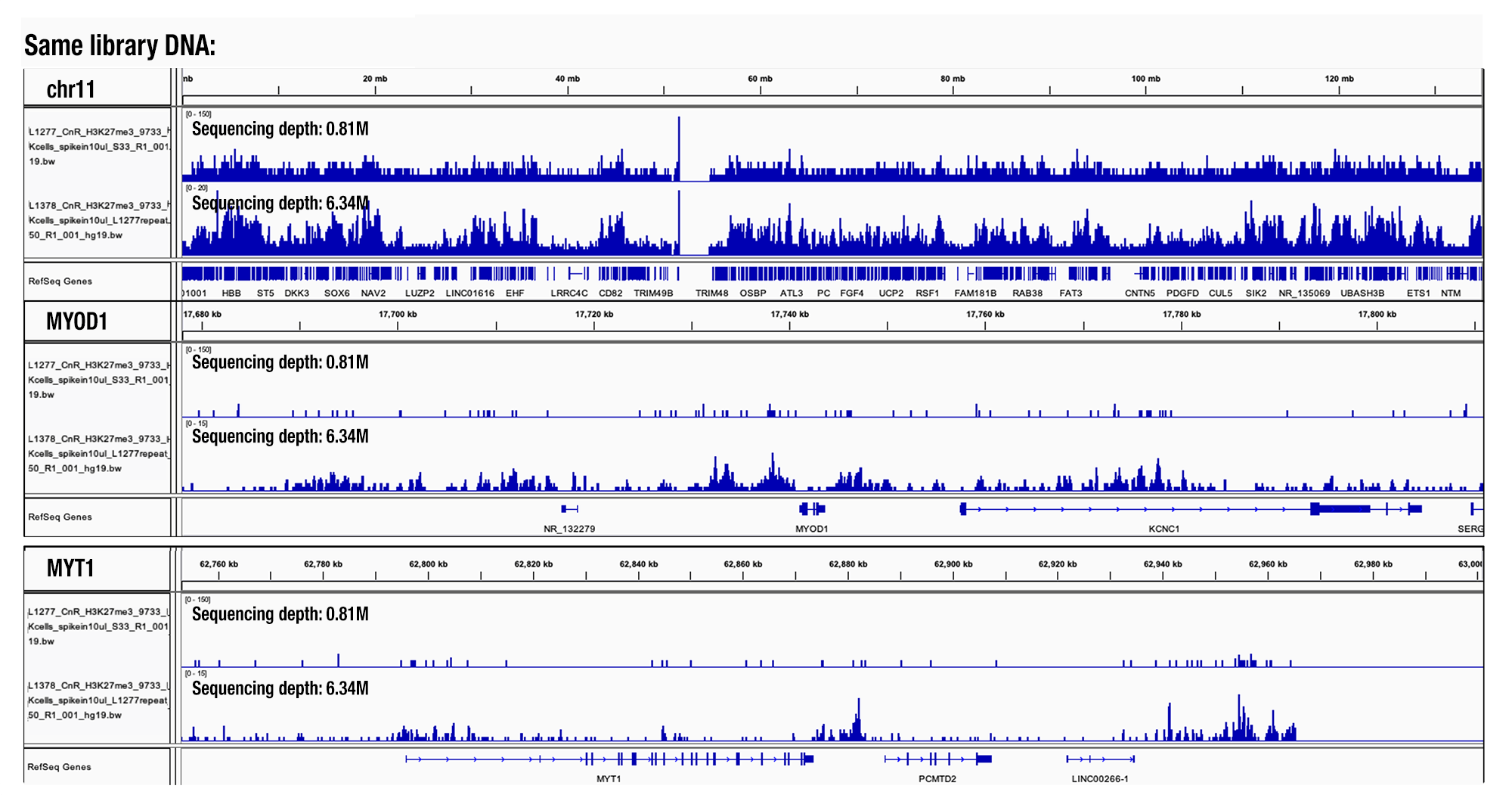
Sequencing Depth Comparison, Target H3K27me3: CUT&RUN was performed with HeLa cells and Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb #9733, using CUT&RUN Assay Kit #86652. DNA libraries were prepared using DNA Library Prep Kit for Illumina (ChIP-seq, CUT&RUN) #56795. Same library DNA were sent for NGS at different sequencing depth as indicated. The figure shows binding across chromosome 11 (top tracks), MYOD1 gene (middle tracks) and the MYT1 gene (bottom tracks).
For any additional questions, please contact our Epigenetic Applications group directly at [email protected].
Christopher Fry, PhD
Director of Epigenetics Product Development
Fang Chen, PhD
Group Leader of Epigenetic Applications Team
posted October 2021